Workflow Quickstarts
Prior to following these quickstart instructions:
1. Ensure conda is installed and the MetaWorks environment is activated.
2. If you will be doing pseudogene-filtering, then ORFfinder needs to be installed.
3. If you are using a custom RDP classifier, ensure it is installed.
If you are unsure of how to do any of this, please refer to the tutorial for step-by-step instructions.
Default ESV Workflow Quickstart
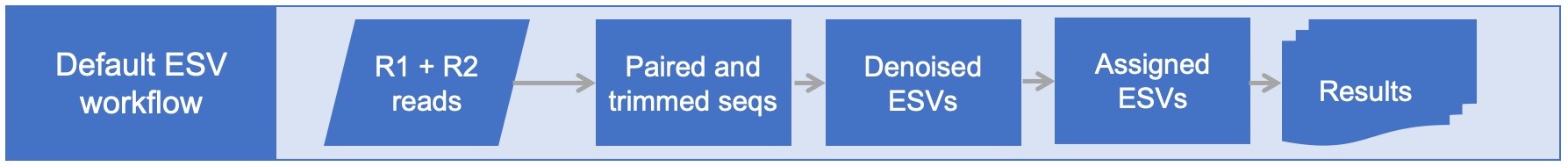
This workflow starts with Illumina paired-end demultiplexed fastq files and generates taxonomically assigned exact sequence variants (ESVs). An adapters.fasta file is required to identify the forward and reverse primers to remove. An example is available in /testing/adapters.fasta . Note that the reverse primer should be reverse-complemented in the adapters.fasta file. Multiple primers sets for the same marker gene can be processed at the same time, E.g. COI_BR5, COI_F230R, COI_mljg. If any of these amplicons are nested within each other, then the primers should be 'anchored' in the adapters.fasta file, see /testing/adapters_anchored.fasta . Different marker genes should be processed separately, E.g. COI, ITS, rbcL.
# quickstart default ESV pipeline
snakemake --jobs 24 --snakefile snakefile_ESV --configfile config_ESV.yaml
OTU Workflow Quickstart
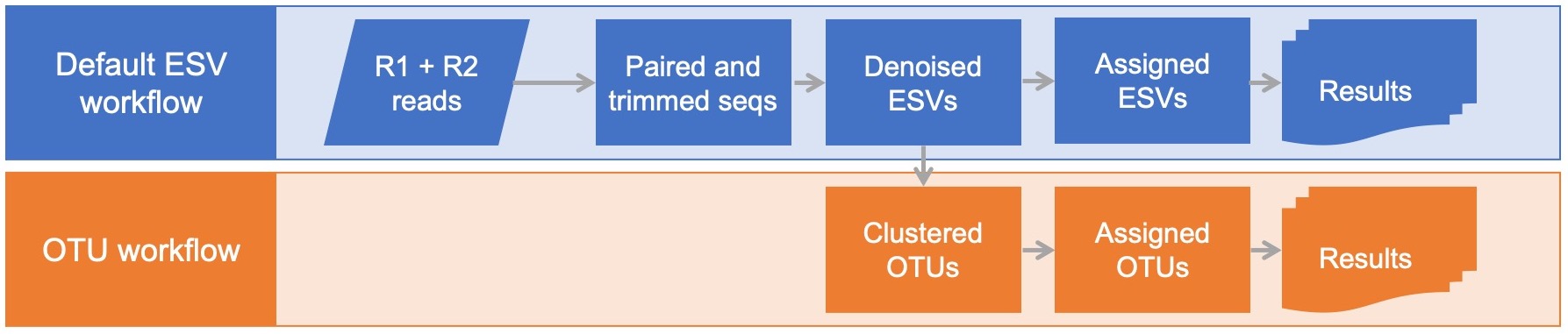
This OTU workflow starts with the taxonomically assigned ESVs from the default dataflow and generates operational taxonomic units (OTUs) based on 97% sequence similarity.
# quickstart OTU pipeline
snakemake --jobs 24 --snakefile snakefile_OTU --configfile config_OTU.yaml
Single Read Workflow Quickstart
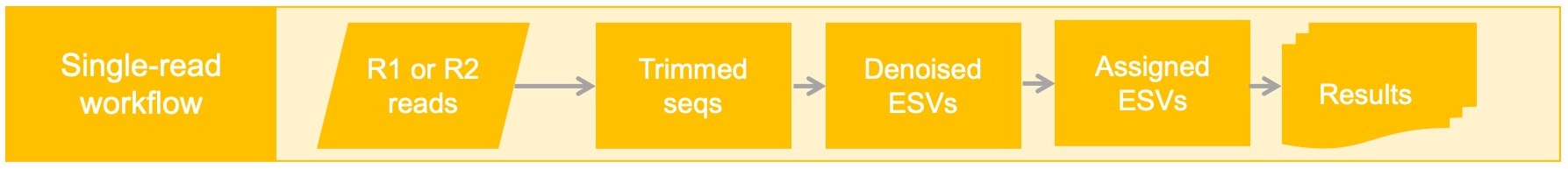
This workflow starts with Illumina paired-end reads that do NOT to overlap. If you would like to process the R1 and R2 files separately this pipeline can be used with caution. Note that if your reads overlap, they should be processed using the default pipeline because longer sequences tend to make better taxonomic assignments with higher accuracy.
# quickstart single read pipeline
snakemake --jobs 24 --snakefile snakefile_ESV_singleRead --configfile config_ESV_singleRead.yaml
Global ESV Workflow Quickstart
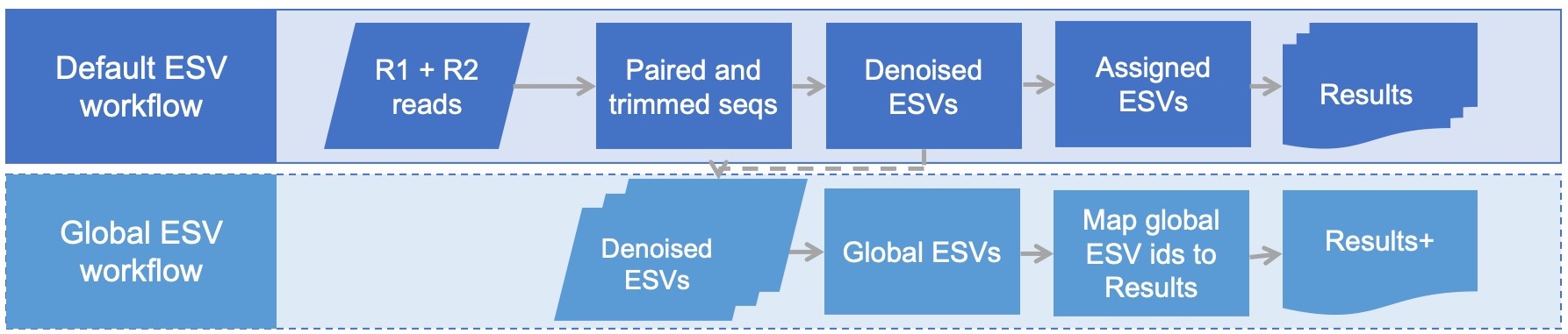
This workflow starts with the taxonomically assigned ESVs from the default dataflow and generates a GLOBAL set of ESV IDs consistent accross all samples *sequenced at different times* to which all ESVs will be mapped. This script may be useful when it is ideal to bioinformatically process samples one season at a time (or one trial at a time, or one year at a time) but still have a consistent set of equivalent ESV IDs project-wide to facilitate multi-season (or multi-trial, or multi-year) comparisons in downstream analyses.
# quickstart global ESV pipeline
snakemake --jobs 24 --snakefile snakefile_ESV_global --configfile config_ESV_global.yaml
Global OTU Workflow Quickstart
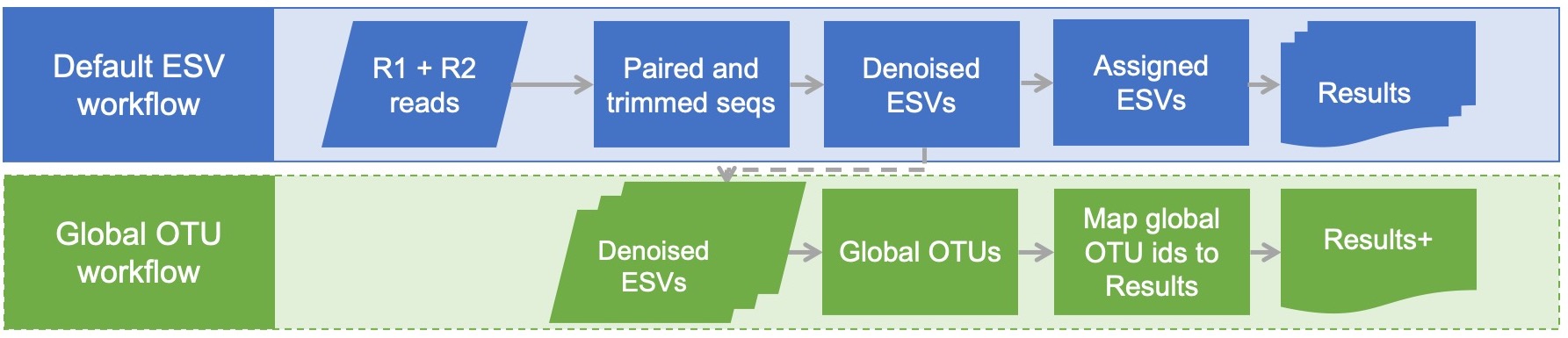
This workflow starts with the taxonomically assigned ESVs from the default dataflow and generates a GLOBAL set of OTU IDs consistent across all samples *sequenced at different times* to which all denoised ESVs willb e mapped. This script may be useful when it is ideal to bioinformatically process samples one season at a time (or one trial at a time, or one year at a time) but still have a consistent set of equivalent OTU IDs project-wide to facilitate multi-season (or multi-trial, or multi-year) comparisons in downstream analyses.
# quickstart global OTU pipeline
snakemake --jbos 24 --snakefile snakefile_OTU_global --configfile config_OTU_global.yaml
Barcoding Workflow Quickstart
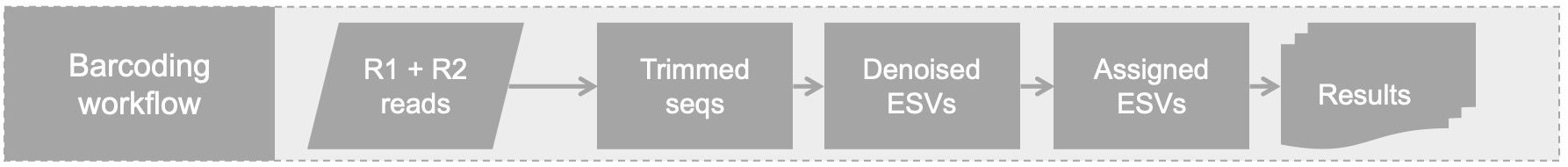
This workflow can handle dual-indexed amplicons sequenced from individuals (as opposed to bulk samples) that were pooled prior to Illumina indexing. An extra mapping file is needed to sort out individuals and amplicon(s) and should contain the following fields: SampleID, Amplicon, Forward, Reverse. This should be saved as a comma-separated values (csv) file. An example of a DualIndexedSamples.csv is available in the 'testing' directory. The SampleID column should contain the individual number associated with a particular dual-index combination. The Forward column should contain the 5'-TagForwardPrimer sequence. The Reverse column should contain the 5'-TagReversePrimer sequence. In our experiments, tags ranged from 5 - 6 bp. The total TagPrimer length ranged from 25-27bp. For maximum specificity, the cutadapt -O parameter in the configuration file should be set to the minimum TagPrimer length range, ex. 25bp in our example.
# quickstart dual indexed sample pipeline
snakemake --jobs 24 --snakefile snakefile_dualIndexedSamples --configfile config_dualIndexedSamples.yaml