Pipeline Details
ESV Workflow
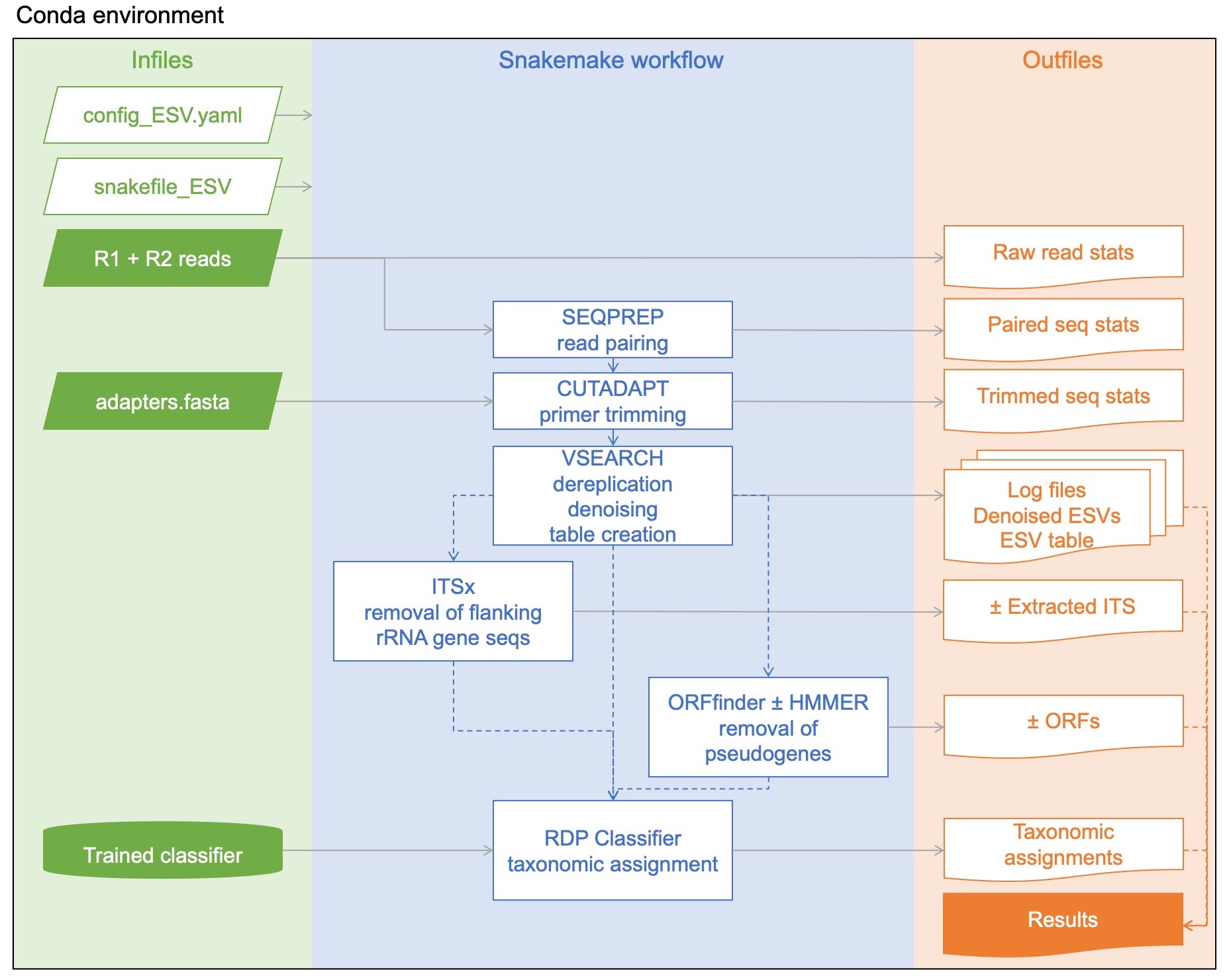
Raw paired-end reads are merged using SEQPREP v1.3.2 from bioconda (St. John, 2016). This step looks for a minimum Phred quality score of 13 in the overlap region, requires at least a 25bp overlap. These paramters are adjustable. Using Phred quality cutoff of 13 at this step is actually more stringent than using a Phred quality score cutoff of 20 at this step as more bases will exceed the cutoff when aligning the paired reads and more mismatches (if present) are counted.
Primers are trimmed in two steps using CUTADAPT v4.1 from bioconda (Martin, 2011). This step now uses the linked adapter approach to remove forward and reverse primers in one step. Primer sequences need to be specified in an adapters.fasta file and the user may wish to anchor them or not, see the CUTADAPT manual for details. At this step, CUTADAPT looks for a minimum Phred quality score of at least 20 at the ends, no more than 10% errors allowed in the primer, no more than 3 N’s allowed in the rest of the sequence, trimmed reads need to be at least 150 bp, untrimmed reads are discarded. Each of these parameters are adjustable.
Files are reformatted and samples are combined for a global analysis.
Reads are dereplicated (only unique sequences are retained) with VSEARCH v2.21.1 from bioconda using the fastx_uniques method (Rognes et al., 2016).
Zero-radius OTUS (Zotus) are generated using VSEARCH with the unoise3 algorithm (Edgar, 2016). They are similar to OTUs delimited by 100% sequence similarity, except that they are also error-corrected and rare clusters are removed. Here, we define rare sequences to be sequence clusters containing only one or two reads (singletons and doubletons) and these are also removed as ‘noise’. We refer to these sequence clusters as exact sequence variants (ESVs) for clarity. For comparison, the error-corrected sequence clusters generated by DADA2 are referred to as either amplicon sequence variants (ASVs) or ESVs in the literature (Callahan et al., 2016). Putative chimeric sequences are then removed using the uchime3_denovo algorithm in VSEARCH.
An ESV x sample table that tracks read number for each ESV is generated with VSEARCH using –search_exact.
For ITS, ITSx v1.1.3 ITS extractor is used to remove flanking rRNA gene sequences so that subsequent analysis focuses on just the ITS1 or ITS2 spacer regions (Bengtsson-Palme et al., 2013).
The standard pipeline (ideal for rRNA genes) performs taxonomic assignments using the Ribosomal Database classifier v2.13 (RDP classifier). We have provided a list of RDP-trained classifiers that can be used with MetaWorks.
If you are using the pipeline on a protein coding marker that does not yet have a HMM.profile, such as rbcL, then ESVs are translated into every possible open reading frame (ORF) on the plus strand using ORFfinder v0.4.3 (Porter and Hajibabaei, 2021). The longest ORF (nt) is reatined for each ESV. Putative pseudogenes are removed as outliers with unusually small/large ORF lengths. Outliers are calcualted as follows: Sequence lengths shorter than the 25th percentile - 1.5*IQR (inter quartile range) are removed as putative pseudogenes (ore sequences with errors that cause a frame-shift). Sequence lengths longer than the 75th percentile + 1.5*IQR are also removed as putative pseudogenes.
If you use the pipeline on a protein coding marker that has a HMM.profile, such as COI arthropoda, then the ESVs are translated into nucleotide and amino acid ORFs using ORFfinder, the longest orfs are retained and consolidated. Amino acid sequences for the longest ORFs are used for profile hidden Markov model sequence analysis using HMMER v3.3.2. Sequence bit score outliers are identified as follows: ORFs with scores lower than the 25th percentile - 1.5*IQR (inter quartile range) are removed as putative pseudogenes. This method should remove sequences that don’t match well to a profile HMM based on arthropod COI barcode sequences mined from public data at BOLD.
The final output file is results.csv. Using report_type 1, it lists each ESV from each sample, with read counts, and includes ESV/ORF/trimmed ITS sequences. Using report_type 2, key files are listed in results.csv but the ESV.table, taxonomy.csv, and FASTA-formatted sequences (ESVs/ORFs/trimmed ITS) are kept separate. Additional statistics and log files are also provided for each major bioinformatic step.
OTU Workflow
If you choose to further cluster denoised ESVs into OTUs, then this is done using the cluster_smallmem method in VSEARCH with id set to 0.97. Primer trimmed reads are then mapped to OTUs to create an OTU table using the usearch_global method with id set to 0.97. The remainder of the pipeline proceeds as described above. The final output file is results_OTU.csv.
Single Read Workflow
If you are using the single read pipeline, the read pairing step with SeqPrep is skipped. If processing the R1 read, raw read statistics are calculated, then the primer is trimmed using CUTADAPT as described above. If processing the R2 read, raw read statistics are calculated then the read is reverse-complemented before the primer is trimmed using CUTADAPT as described above. The final file is results.csv. When assessing the quality of your taxonomic assignments, be sure to use the appropriate bootstrap support cutoffs for these shorter than usual sequences.
Global ESV Workflow
If you have samples sequenced at diffrent times (multiple seasons, years, or trials), you will likely process these samples right after sequencing resulting in multiple sets of ESVs. To facilitate downstream processing, it may be advantagous to have a GLOBAL set of ESV ids so that data can be compared at the ESV level accross seasons, years, or trials. The directories containing the output files processed using the default pipeline need to be in the same directory as the snakefile script. Ex. 16S_trial1, 16S_trial2, 16S_trial3. In each of these directoreis, there is a cat.denoised.nonchimeras file and a results.csv file. The denoised (chimera-free) ESVs (or ITSx processed ESVs) are concatenated into a single file, dereplicated, relabelled using the SHA1 method. This file then becomes the new global ESV sequence database. A fasta file is generated from the results.csv file and these sequences are clustered with the new global ESV sequence database using the usearch_global methods with the id set to 1.0. The global ESV that each trial ESV clusters with is parsed and mapped to the final output file called global_results.csv.
Global OTU Workflow
If you have samples sequenced at diffrent times (multiple seasons, years, or trials), you will likely process these samples right after sequencing resulting in multiple sets of ESVs. To facilitate downstream processing, it may be advantagous to have a GLOBAL set of OTU ids so that data can be compared at the ESV level accross seasons, years, or trials. The directories containing the output files processed using the default pipeline need to be in the same directory as the snakefile script. Ex. 16S_trial1, 16S_trial2, 16S_trial3. In each of these directoreis, there is a cat.denoised.nonchimeras file and a results.csv file. The denoised (chimera-free) ESVs (or ITSx processed ESVs) are concatenated into a single file, dereplicated, relabelled using the SHA1 method, then clustered into OTUs with 97% sequence similarity. This file then becomes the new global OTU sequence database. A fasta file is generated from the results.csv file and these sequences are clustered with the new global OTU sequence database using the usearch_global methods with the id set to 0.97 . The global OTU that each trial ESV clusters with is parsed and mapped to the final output file called global_results.csv.
Barcoding Workflow
If you are sorting out tagged individuals, it is advised to set the CUTADAPT -O parameter to maximize the tag+primer overlap with the read. For example, if the range of tag+primer lengths is 25 - 27bp, then set -O 25 (default is 3 bp). If the tags are short, then it is especially important to ensure no mismatches between this and the read. For this reason, it is recommended to set the CUTADAPT -e parameter to allow for zero errors between the tag+primer and read, -e 0 (default is 0.1 or 10% errors).